




I. 서론
과거 터스키기 사건과 같이 연구 진행 과정에서 윤리에 소홀을 초래한 전례들은 연구의 윤리적 수행과 준수의 중요성을 일깨워줬고, 그 결과가 연구자, 연구대상자 그리고 국가적으로 큰 손실을 야기할 수 있음을 알려줬다. 우리 역시 황우석 사태를 겪으며 생명윤리와 안전에 대한 중요성을 경험한 바 있다. 과거 경험을 토대로 「생명윤리 및 안전에 관한 법률[1](이하 ‘생명윤리법’)」은 2012년 2월 전면개정을 통해 규제 영역을 확대하고, 기관생명윤리심의위원회를 배아 및 유전자연 구뿐 아니라 인간 및 인체유래물연구에 대한 연구계획서 등의 심의와 교육, 조사·감독까지 수행 하는 연구 관련 총체적 관리 기구로 규정하며 연구의 윤리와 안전성 확보를 위해 노력해왔으나, 연구 수행에서 윤리를 간과한 문제는 여전히 발생한다. 예컨대, 언론을 통해 종종 동의 또는 보고 절차를 미준수[2,3,4]1)하거나 취약한 연구대상자를 대상으로 부적절하게 연구를 수행한 사례[5]가 보도되기도 했고, 최근에는 정치 관계자 자녀들이 기관생명윤리위원회(이하 ‘기관위원회’ 또는 ‘IRB’) 승인을 허위로 기재하거나 연구 수행 전 기관위원회로부터 승인을 받지 않은[6,7] 사례들이 발생하였다. 일련의 스캔들로 연구의 윤리적 수행에 대한 중요성이 부각되고 있음에도 불구하고, 이러한 사건·사고는 발생 이후 잠시 주목을 받고 관리 방안의 필요성이 대두되다가 명확한 대안 없이 잊히는 과정을 반복해왔다. 지금까지 생명윤리법을 통해 생명윤리와 안전을 확보하는 연구 체계를 수립하고 제도를 활성화하는 데 전력을 쏟았다면, 이제는 법이 적절히 준수되지 않았을 때의 관리 방안과 이를 예방할 수 있는 안전장치는 무엇 일지로 초점을 옮겨 구체적인 방안을 강구해야 할 때이다. 관리 체제의 결여는 연구의 윤리성과 신뢰성을 책임져야 하는 해당 기관과 기관위원회의 기반을 위협하고 각종 연구윤리 문제가 되풀이되도록 하기 때문이다. 사회 변화와 과학기술발전이 그 어느 때보다 급격한 현재, 연구 환경에서 발생 가능한 윤리적 이슈는 더욱 다양하고 위협적일 수 있으며, 과거의 과오를 되풀이하지 않기 위해서라도 연구 관련 발생 가능한 윤리적 문제와 이를 준수하지 않을 때 관리 방안의 고려가 필요하다.
인간을 대상으로 하는 연구의 윤리적 수행은 일 반적으로 연구대상자의 권리나 안전, 복지 등의 보호를 위해 필요하다고 판단되며, 기관위원회가 승인한 범위(승인된 연구계획서)를 적절하게 준수하지 않은 경우를 연구 미준수로 보고 이를 관리 한다. 본 글에서는 심의 대상이지만 연구자가 기관위원회의 심의를 받지 않고 연구를 수행하거나, 결과에서 심의 등 윤리적 의무(기관위원회의 승인)를 허위로 기재하는 등의 사항을 심의 미준수로 정의하고 이에 대한 관리 방안을 모색하고자 한다. 현재 생명윤리법은 심의 의무를 규정하고 있지만 실제 그 적절한 준수 여부가 관리되지 않아 심의 미준수가 양산되는 측면이 있으므로 연구자의 심의 의무를 강화할 수 있는 예방적, 정책 및 제도적 관리 방안을 마련하는 것이 필요하다. 또한 IRB 심의는 연구의 윤리적 시작과 안전은 물론 진행과 종료를 담보하는 과정으로, 심의 관련 미준수를 제도적으로 관리하는 방안 마련은 중요할 것이다. 이에, 본 글은 연구 수행과 관리에 대한 생명윤리법의 취지와 한계를 먼저 살펴보고, 우리와 유사하게 기관 내 IRB를 설치·운영하는 미국의 IRB 심의 의무와 관리에 대한 법제를 검토하고자 한다. 그리고 연구의 시작과 결과 관리와 관련된 연구 지원 및 학술 기관의 정책과 IRB 관리 정책을 살펴봄으로써 IRB 심의 준수 관리 방안에 대한 시사점을 도출하고자 한다.
II. 연구에 대한 생명윤리법의 기본 방향 및 한계
현재 의과학과 사회과학연구를 포괄한 연구의 기관위원회 심의와 연구 수행·관리를 규제하는 법률은 「생명윤리 및 안전에 관한 법률(이하 ‘생명 윤리법’)」이다. 2004년 제정된 생명윤리법은 생명 윤리와 안전의 확보가 필요한 분야를 인간의 배아, 세포, 유전자로 규정하고, 기관생명윤리심의 위원회를 통해 배아 및 유전자 관련 연구계획서나 동의 및 개인 정보보호 등을 심의하도록 하였다. 이후 생명윤리와 안전의 가치가 확보되어야 하는 분야가 인간이나 인체유래물등을 이용하는 연구로 확대되는 연구 환경의 변화에 따라, 2012년 전 면개정을 통해 적용 범위를 “인간과 인체유래물 등을 연구하거나 배아나 유전자 등을 취급할 때”로 확대하고, 기관생명윤리위원회의 설치 의무를 법 제10조에 따른 대학 및 연구기관 등으로 확장하였다. 또한 위원회 역할을 연구계획서의 동의, 안전 등에 대한 심의뿐 아니라, 연구 수행 과정 및 결과에 대한 조사·감독과 연구자 교육, 윤리지 침 마련, 취약한 연구대상자 등의 보호대책을 수립하도록 강화하여, 기관에서 발생 가능한 윤리적 문제, 안전과 위험을 위원회를 중심으로 기관 스스로 관리하는 자율규제 방식을 택했다. 전면 개 정된 법의 궁극적 취지는 연구자와 기관의 윤리적 인식 개선과 강화를 통해 자율성에 근거한 책임 있는 연구 수행을 강조한 것이다.
이처럼 기관 내 생명윤리 및 안전의 확보가 자율적으로 이루어지기 위해서는 ‘기관-연구자-기관위원회’ 간 유기적 상호 연계가 필수적이다. 즉, 기관은 기관위원회 운영에 필요한 자원과 독립성 보장을 지원해야 하며, 연구자는 본인이 수행하는 연구와 관련한 의무와 책임을 인지하고 준수해야 하고, 위원회는 기관 내 생명·안전·윤리 관리 조직이라는 위상 하에 연구자를 교육하고 연구자의 연구 행위를 관리·감독해야 한다. 하지만 일부 기관은 연구 수행과 관리를 자율적으로 규제 하는 데 어려움을 겪기도 한다. 기관위원회가 연구에 대한 관리 조직으로 적절한 역할을 수행하기 위해서는 기관으로부터 지원이 필요하며, 특히 기관위원회 운영을 지원하는 전담인력의 지원은 필수적이다. 하지만 대부분의 기관에서 기관위원회 운영지원인력은 전담이 아니거나 잦은 인사이동으로 업무의 연속성과 전문성을 보장받지 못하고 있다[8]. 기관위원회 위원도 심의 및 관리를 위해 적절한 대우와 지위가 필요하나 기관으로부터 지원 확보가 어렵다[9]. 연구자 관리 측면에서 기관위원회의 위상을 보여주는 조사·감독의 경우, 조사·감독 기준은 마련하고 있으나 실제 조사·감독을 시행한 이력이 없거나 규정에 따른 후속 조치 절차를 준수하지 않은 경우가 대부분이다. 이러한 상황은 아직 현장에서 기관위원회가 연구 관리를 위해 기관으로부터 충분한 지원도, 연구자에게는 연구의 조사·감독을 수행할만한 굳건한 위 상이 부족하여 연구의 윤리성과 안전성 확보를 위한 생명윤리법의 자율규제가 어려울 수 있다는 바를 간접적으로 보여준다.
기관위원회의 업무 범위도 한계로 작용한다. 생명윤리법은 인간대상연구 및 인체유래물연구자에게 심의의 의무를 부여한다. 따라서 연구자가 연구 수행 전 IRB 심의를 받아 의무를 수행하면, 기관위원회는 해당 연구를 관리하고 기관은 기관위원회의 운영을 지원하는 이상적인 체제를 갖춘다. 하지만 그 출발점에 놓인 연구자가 심의 의무를 준수하지 않을 때는 이러한 관리 체계가 더는 작동되기 어렵다. 연구를 IRB 심의를 받은 연구와 그렇지 않은 연구로만 구분한다면, 기관위원회가 관리할 수 있는 연구 범위는 IRB 심의를 받은 연구로 한정되기 때문이다. 기관위원회는 생명 윤리법 제10조제3항에 따라 심의, 조사·감독, 교육, 취약한 연구대상자 보호 대책 수립과 지침 마련 등을 수행해야 하나, 연구계획서를 제출하지 않은 연구에 대해서는 심의는 물론, 조사·감독도 시행이 어렵다. 또한 연구계획서가 처음 제출되었을 때 신규 심의를 시작으로, 지속·변경·종료보고에 대한 심의를 통해 연구의 윤리성, 과학성, 동의, 안전 등을 확인하므로 애초 심의가 신청되지 않은 연구는 관리가 불가능하다.
생명윤리법은 연구의 윤리적 수행과 안전을 유지하기 위한 최소한의 약속이며, 법의 특성상 처벌과 규제 범위가 세세할수록 행동에 제약과 자유에 제한이 발생한다. 법을 아무리 촘촘히 만든다고 하더라도 사각지대 발생을 완전히 막을 수는 없으므로 IRB 심의를 준수하지 않은 연구의 관리를 법적 제재를 통해 해결하려는 바는 적절하지 않다고 본다. 이러한 상황과 관점에 비추어, 연구 관리에 대한 생명윤리법의 자율규제가 적절히 구현되기 위해 필요한 부분은 무엇일지, 현재의 자율규제 방식을 유지하되 IRB 심의를 받지 않고 수행된 연구를 관리할 방안이 있는지, 제도적으로 보완될 부분이 있을지에 대한 검토가 필요하다.
III. 국내와 미국의 IRB 심의 관련 법제와 정책 비교
IRB 심의 이행과 준수에 대한 관리는 법률과 더불어, 연구 관련 기관 정책과도 긴밀히 연계된다. 이에 본 장에서는 우리나라와 미국의 비교를 통해 IRB 심의 의무와 관리를 규정한 법제 검토를 시작으로, 심의를 담당하는 IRB에 대한 관리 정책과 연구 관리 기관 및 학술 기관의 관리 정책을 차례로 살펴보고자 한다.
생명윤리법은 제15조(인간대상연구의 심의)와 제36조(인체유래물연구의 심의)에서 해당 연구를 수행하는 자는 연구 수행 전 연구계획서를 기관위원회에 제출하고 심의받아야 한다고 규정한다. 심의를 제출하고 받아야 하는 자는 연구자로 IRB 심의는 연구자의 법적 의무다. 이에 대한 관리는 법 제14조에 따라 기관위원회의 구성과 운영을 평가하여 인증하는 형태로 수행되며, 인증 결과에 따라 중앙행정기관의 장은 기관에 예산 지원과 국가 연구비 지원 제한 등의 조치를 취할 수 있다. 또한, 생명윤리 및 안전에 중대한 위해가 발생하거나 발생할 우려가 있음에도 기관장이 IRB 소집 또는 보고 등의 의무를 하지 않은 경우 기관에 과태료를 부과할 수 있도록 규정한다. 즉, 생명윤리법에서 IRB 심의 의무는 ‘연구자’에게 있으나, 심의 준수는 ‘기관위원회’가 관리하고, 위반 시 제재는 ‘기관’이 받도록 하여, 기관 구성원들 스스로가 활동을 자율적으로 규제하는 제도를 정착시키고자 했다. 하지만 앞서 살펴본 바와 같이 연구자가 심의 의무를 준수하지 않은 경우에는 이러한 관리 체계의 작동이 어렵게 되며, 현실적으로도 연구자의 심의 의무와 준수는 적절히 관리되고 있지 못하다.
우리와 유사하게 기관 내 IRB 설치와 운영을 통해 연구를 관리하는 미국은 보건부(Department of Health and Human Services) 연방 규정 45CFR46[10]을 통해, 연방 부서 또는 연방 기관이 수행하거나 지원하는 인간대상연구를 관리한다. 해당 규정은 보건부 규정이나 총 18개 부처가 채택하여 따르는 커먼룰(common rule)로 미국 내 연구대상자를 보호하고 관리한다. 45CFR46의 제 46.103조(준수 보증)는 심의면제 대상(제46.104 조)을 제외한 모든 연구를 심의 대상으로 규정하며, 해당 연구를 수행하는 자는 IRB 심의 의무를 갖는다. 또한 기관에 본 규정을 준수한다는 보장 (assurance)에 서약하도록 하고 기관 내 모든 심의 대상 연구가 IRB 심의와 승인 하에 수행한다는 증명서(certification)를 제출하도록 정한다. 이로써, 연구자가 IRB 심의를 받아야하는 의무뿐 아니라, 기관에게도 연구자의 IRB 심의 의무를 감독하고 준수를 보장하는 책임 권한을 부여한다. 기관이 제출한 보증 서약과 IRB 심사·승인을 입증하는 증명은 보건부 산하에 인간대상연구보호국(Office of Human Research Protection, 이하 OHRP)2)에서 검토한다. OHRP는 서약과 증명을 충족 한 기관과 연방차원의 질보증 확약(federalwide assurance, 이하 FWA)을 맺어 관리한다. 보증 서약과 증명이 승인되기 전 연구는 시작될 수 없으며, 해당 기관이 IRB 심의 준수를 증명하지 못한 경우에는 연구비 지급이 중단될 수 있다. 국내와 미국의 IRB 심의 관련 법제는 다음과 같이 정리될 수 있다.(<Table 1> 참조)
종합하면 생명윤리법과 미국의 45CFR46 모두 법적으로 해당 연구를 수행하는 연구자에게 IRB 심의 의무가 부여되는 것은 동일하다. 다만, 이를 관리할 때 생명윤리법은 기관위원회의 구성 및 운영실적 등을 평가하므로 기관위원회가 심의한 실적을 중심으로 심의 의무 등이 소극적으로 평가된다. 즉, 심의를 신청하여 관리하고 있는 기관위원 회의 역할 및 운영의 적절성을 위한 평가에는 적절하지만 보다 근본적으로 기관 내 종사자 또는 연구자가 생명윤리법을 얼마나 준수하고 있는지에 대해 관리로는 적절하지 않다. 이를 보완하기 위해 2016년부터 시행하고 있는 기관위원회 평가 시 적용하는 기준이 있으나 그 실효성을 기대하기는 어렵다는 지적이 있다.3) 반면, 미국은 기관에 실제 IRB 운영과 연구 수행과 준수 의무를 포괄적으로 관리하도록 요구하고 이를 근거로 기관에게 연구비 지급 중단 등의 제제를 할 수 있도록 하여, 결과적으로 기관이 연구자의 법률 미준수를 관리 할 책임을 강조하는 효과를 갖게 된다. 연구자의 미준수를 누가, 어떤 방식으로 관리하는 것이 효율적일지 또는 바람직할지에 대해서는 다양한 논란이 있을 수 있고 실제 연구자의 책임이 가장 기본이 되어야 하겠으나, 이를 효율적으로 관리하고 무엇보다 실질적으로 관리 가능하도록 기관 또는 기관위원회에 책임과 관리 영역을 명확히 하는 관리 방안의 제시가 중요할 것이다.
기관위원회가 연구자의 IRB 심의 준수를 확인 하고 관리하기 위해서는 연구자를 교육하고 연구 행위를 관리·감독할 수 있는 역량이 필수적이다. 법에 따라 IRB 스스로가 기관 내 연구자 및 종사자를 교육하고 연구자들의 역량을 개발·강화해야 할 의무가 부여되어 있으나, IRB별로 적절한 교육을 자체적으로 개발하여 제공하거나 연구자를 전적으로 관리하기에는 현실적으로 한계가 있다. 따라서 IRB를 관리하는 정부나 유관기관으로부터 지원이 요구된다. 현재 국내 IRB는 업무와 역할에 따라 각기 다른 기관으로부터 관리와 지원을 받는다.
기관위원회 등록은 질병관리본부가 관리하며, 교육은 보건복지부 고시[11]에 따라 위원 교육은 대한기관윤리심의기구협의회(이하, KAIRB)에 지 정되어 있다. 그 외에도 한국보건복지인력개발원, 질병관리본부, 국가과학기술개발원 및 공용 IRB 등에서 생명윤리법에 따른 기관위원회 또는 연구자를 위한 다양한 교육을 제공하고 있다. 등록된 기관위원회들에 대한 구성 및 운영실적을 평가하는 기관위원회 평가·인증은 국가생명윤리정책원에 위탁되어 있다. 생명윤리법은 기본적으로 기관위원회에 의한 자율적 운영에 기반하나, 운영 현황에 대한 조사나 교육, 평가 및 인증 등에 관한 규정을 마련한 것은 자율적 운영에 대한 어느 정도의 간접적 질 관리를 염두에 둔 것이다. 따라서 기관위원회가 적절한 역량을 가지고 역할을 수행하도록 하는 것이 무엇보다 중요할 것이며, 이를 위해서는 현재의 분절적 관리보다는 단계별 일관된 정책을 통해 기관위원회를 보다 효율적으로 관리 및 지원할 필요가 있다. 특히, 현재 기관위원회 등록은 IRB 현황 관리를 위해 마련된 제도이나 등록 정보에 대한 실질적 관리가 되지 않아 정보의 효율성이 낮다는 지적이 있다. 교육의 경우, 임상시험 연구자는 1995년부터 시행된 「임상시험관리기준」과 2017년 「의약품 임상시험 등 종사자 교육 및 교육 실시기관 지정에 관한 규정」에 따라 대상자별 표준화된 교육 과정을 통해 관리되고 있으나, 대학 등 비임상연구 분야의 연구자, IRB 위원 또는 행정간사들에 대한 교육은 교육 정책이나 콘텐츠가 일관성 있게 조절되거나 관리되고 있지는 않다. 여러 기관에서 일원화되지 않은 다양한 교육4)을 받고 있어, 대학이나 연구기관 등에서 수행되는 연구에 대한 이해를 바탕으로 한 적절한 교육과 안내가 필요하다는 지적도 있다[8,12,13].
또한 현재 시행되고 있는 기관위원회 평가제도는 기관위원회 운영의 적절성을 평가하면서 기관위원회의 조사·감독이 적절히 운영되고 있는지를 평가하고 있으나, 대부분의 기관위원회 표준 운영지침서(SOP)는 조사·감독 대상 범위를 IRB 심의를 받고 수행 중인 연구로 한정하고 있다. 실제 법은 “해당 기관에서 수행되는 연구”라고 포괄적으로 기술하여 심의 여부와 관계없이 조사·감독의 대상이 될 수 있도록 하였으나, 기관에서 현실적으로 연구 시작 전 심의 의무를 관리하거나, 심의 받지 않은 연구에 대하여 조사·감독 등을 수행하는 것은 어렵다고 판단하여 제외하고 있다. 이러한 한계를 인지하여 2016년부터 시행되는 평가에서는 해당 기관 내 연구자의 심의 의무에 대한 기관위원회의 역할을 확인하기 위해 “기관 내 연구자 및 종사자가 생명윤리법에서 규정하는 의무를 이행하는지 여부를 관리한다.”라는 평가기준을 마련하여, 해당 기관에서 이미 수행된 국책과 제 중에서 심의 대상일 가능성이 높은 과제를 추정하여 심의 준수 여부를 확인하는 방식으로 검토하고 있다. 하지만 이때 IRB 심의 대상 여부에 대한 최종 확인은 해당 과제를 수행한 연구자들의 답변에 의존하고 있어, 연구자의 심의 미준수에 대한 관리 여부를 추정할 수 있을 뿐 평가 목적에 따른 실효성을 기대하긴 어렵고, 구체적 근거가 없어 현실적이지 않다고 지적된다. 인증제 도입 전 등록된 기관위원회 전수를 대상으로 기관위원회 전반의 수준을 파악하고 운영을 지원하는 데 목적을 둔 현재 평가제도에서는 용인될 수 있는 평가기준이겠으나, 향후 인증을 전제로 한 평가에서는 실효성이 없거나 현실적이지 않은 기준으로 평가되고 인증될 수는 없다. 형식적 절차로서의 평가가 아닌 기관위원회 운영 현황에 대한 적절한 파악과 개선을 통한 역량 강화로 이어질 수 있는 평가에 대한 고려가 필요하다. 이러한 맥락에서 향후 연구자의 IRB 심의 준수 관리 체제와 IRB와 기관의 의무 등에 대한 평가범위와 기준을 명확하게 마련하는 것은 중요해 보인다.
반면, 미국은 보건부 산하에 인간대상연구 보호 국(Office for Human Research Protections, 이하 OHRP)이란 IRB 관리 전담 부처에서 기관위원회를 총괄적으로 관리한다. OHRP는 IRB가 아닌 기관으로부터 연방규정인 45CFR46을 준수하겠다는 법적 효력이 있는(legally binding assurance) 준수 서약서를 받아 연방 보증 확약(Federalwide Assurance, 이하 FWA)을 맺는다. 이후 확약 체결 기관에게 연구대상자보호 업무를 담당할 IRB를 OHRP에 등록하도록 하고, 인간대상자 보호에 대한 정책과 지침을 개발하여 해당 기관의 연구자를 교육하며, 45CFR46 준수 여부를 점검하여 미준수를 관리한다.5) 이에 따라 기관은 적절한 운영 및 관리를 위해 기관 내에 IRB 외에도 필요하다면 추가적인 관리 체계를 마련할 수 있고 기관별 다양한 거버넌스의 형태로 운영이 가능하다. 기관은 그 준수 여부 등 관리에 대해 OHRP가 요구할 경우 자료 제출의 의무가 있으므로 이를 적절하게 관리 및 기록하며, 관리의 대상을 IRB 심의를 받은 연구에만 한정하지 않는다.
더불어 OHRP는 IRB 심의 미준수를 연방규정을 심각하게 위반하여 즉시 보고가 필요한 미준수로 정하여 관리한다[14]. OHRP는 보고된 미준수에 대하여 기관이 취할 조치의 적절성을 즉각 검토하고, 필요시에는 추가되어야 할 시정 조치를 제안하며, 해당 조치가 향후 미준수 예방에 도움이 될지에 관한 의견을 제공한다. 상시적 의견 교류와 컨설팅을 통해 기관위원회는 미준수를 적절히 관리할 수 있는 역량을 배양하게 된다. 또한 OHRP는 기관의 보고와 정기적 감독 외에도 IRB 승인 없이 연구가 수행되었다는 합리적 의심에 따른 민원을 접수받아 검토한다. 민원인이 제보한 의심 사례를 해당 연구의 책임연구자에게 전달하고, 연구자로부터 소명과 개선 계획을 제출하도록 한다. 최종적으로 OHRP는 책임연구자에게 검토 결과와 그에 따른 제재 조치를 포함한 서신을 전달한다.
주목할 점은 미준수 조사 방법에 상관없이 OHRP는 기관 홈페이지의 ‘위반 및 보고’란에 검토된 모든 사례에 대하여 책임 연구자에게 보낸 제재 조치를 포함한 서신 전문을 공개한다는 것이다. 뿐만 아니라, 아직 미해결된 문제와 현재 진행 중인 조사와 관련된 모든 내용을 홈페이지[15]에 공개한다. 이로써, IRB 심의나 승인을 받지 않은 연구에 대해 조치를 취하고6) 해당 정보를 대중에게 공유하여 투명하게 관리한다. 국내와 비교할 때 미국은 OHRP라는 IRB 관리기구를 통해, 기관위원회 등록·교육·기관위원회 운영 관리가 일원화된다. OHRP는 기관으로부터 IRB 심의를 포함한 법령 준수를 확인하여 보증을 부여하고, 연구자와 IRB 책임자를 대상으로 FWA 하에서 요구되는 주요 책임과 의무를 교육한다. 또한 IRB 심의 미준수 관리 조치에 대하여 의견을 교환하고 법에 따른 제재 조치를 집행하며 이를 대중에게 공개한다. 이에 따라 IRB 운영 전반과 연구자의 심의 의무 등의 행위가 법에 부합되도록 관리·지 원하는 체계를 구현하고 있다.
우리나라에서 국가비 지원 연구 관리와 집행을 책임지는 기관은 한국연구재단이다. 재단은 국제적으로 IRB 심의가 중요시되면서 신규 지원 신청 요강 및 연구비 지원 공고문에 IRB 심의 의무화를 명시하기 시작했다[16]. 연구재단의 IRB 심의 의무화 규정은 연구자들에게 기관 내 IRB 존재와 심의 기능을 알리는데 일조한 것은 분명하나, IRB 심의 확인과 관리를 재단이 아닌 연구 수행 기관으로 명시하여 미준수 발생 시 총체적 책임 기구의 역할은 면제하고 있다.7) 따라서 연구자와 연구 기관을 대상으로 IRB 심의 미준수를 관리하고 방 지할 제도적 방안은 여전히 부재하다.
미국은 국립보건연구원(National Institutes of Health, 이하 NIH)이 보건 의료 분야에 대한 기초·응용연구를 수행하고 연구비를 지원하는 기관이다. NIH는 「연구비 정책(NIH Grants Policy Statement)」 제4.1.15.2.조에 따라, 연구비 신청 자는 대상자가 해당 연구에 참여하기 전 OHRP로부터 부여된 기관의 질보증 확약(FWA) 번호와 연구계획서 및 동의서에 대한 IRB 심사 승인 증명서를 제출하도록 한다[17]. 심사 승인 증명서는 승인일을 포함해야 하며 초기 신청서 제출 시 또는 NIH 동료평가 후 최종 승인 검토 단계에서도 제출할 수 있다. 어떤 경우에도 IRB 승인 증명이 충족되기 전 NIH가 지원하는 인간대상연구는 시작될 수 없다. 뿐만 아니라, 연구자는 연구대상자보호와 관련한 변동 사항을 연구 중간 보고 시 보고서에 포함해야 하고 필요시에는 진행 중 연구의 IRB 준수를 보증하는 IRB 지속심사승인서 (Annual IRB Approval)를 제출하도록 정한다[18]. 연구비 수여자들은 연구 진행 문서와 증빙서류를 eRA Commons라는 웹사이트에 제출하고, 미제 출하거나 연구 수행의 적절성이 부적합하다고 판단 시에는 연구비 수여 연기 또는 수여 조건 보류, 박탈 등의 조치가 가능하다.
한국연구재단도 「정책 연구 용역과제 관리지침」 제20조(연구결과의 보고)에 연구 기간 1/2 시점에 중간 보고서를 재단에 제출하도록 하고 있으나, 제출 내용은 ‘과제 개요’, ‘중간 과제 수행 및 절차’, ‘중간 추진 결과’, ‘향후 추진 계획’ 등으로 주로 연구 관련 내용에 한정된다[19]. 연구용역비 지급 중단 및 회수 기준(제18조)의 하나로 “연구 진행 상황 보고를 하지 않거나 허위로 한 경우(제18 조3항)” 또는 “연구부정 행위(표절, 변조 등)에 의한 연구결과를 발표·제출한 경우(제18조제4항)”를 두고 있으나, IRB 심의와 승인에 대한 사항은 본 지침에서 정의하는 부정행위 및 허위사실에 포함되지 않는다.
결과 관리에 대한 정책은 생명윤리법과 미국의 45CFR46 모두 명시적인 규정을 가지고 있지 않으며, 연구윤리 차원에서 관리되고 있다. 그러나 연구의 계획과 시작은 물론 연구의 결과를 활용하는 단계까지 윤리를 준수하는 것은 연구의 완전성(integrity) 측면에서 중요하다. 우리나라의 경우, IRB 심의를 받지 않은 연구결과에 대하여 한국연구재단은 논문 게재 거부 등 학계의 자율적인 제재 조치가 가능하다고 명시하여[20], 이를 통합적으로 관리할 방안이 없다. 따라서 학술지를 발행하는 대부분의 학술기관은 해당 연구자가 IRB 심의를 받았다고 하면 이를 믿고, 학회 차원에서 IRB 승인 번호를 요구하더라도 사실 여부까지는 확인하지 않으며, 문제 발생 시만 해당 논문을 철회하는 등의 형태로만 관리한다. 학술기관은 단순 논문 발간뿐 아니라, 믿을만한 그리고 가치 있는 지식을 공유하여 학문적 가치를 높이고 연구윤리를 함양할 책임도 갖는다는 점을 고려할 때, 문제 발생 시에만 바로잡는 수동적 자세보다는 연구의 윤리적 기초와 수행을 보증하는데 선제적 태도를 취할 필요가 있다.
학술지 정책은 학문 분야별로 국제적 지침을 마련하는 등 해당 학문 분야의 권위 하에서 연구자의 연구결과를 관리한다. 영국은 출판윤리 위원회(Committee on Publication Ethics, 이하 COPE)를 통해 출판 윤리를 관리하고 있다. COPE는 저널 편집자들이 연구 부정행위 의심사 례(allegations of misconduct)에 직면했을 경우 활용 가능한 사례별 관리 흐름도(flowchart)를 제시한다. 특별한 점은 일반적으로 연구 부정행위에 포함되는 위조, 날조, 변조 외에 윤리적 문제까지도 포함한다는 것으로, 논문 심사자가 윤리적 승인이 필요한 연구임에도 절차를 거치지 않다고 판단하거나 동의 및 연구대상자 보호와 관련하여 윤리적 우려를 제기할 때 부정행위 관리 절차를 밟도록 제안한다[21,22].
국제 의학학술지의 규정, 강령, 편집, 발간에 대한 권장사항을 협의하는 국제의학학술지 편집인 협회(International Committee of Medical Journal Editors, 이하 ICMJE)는 「의학학술지 학술 업적의 수행, 보고, 편집, 발간에 관한 권고 지침」[23]을 발간하여 의학학술지 운영의 모범사례를 설정하고 윤리 표준을 제안한다. 지침 E.(연구대상자 보호)는 모든 연구자는 「헬싱키 선언」[24]에 따라 인간대상연구의 수행과 보고가 이루어졌는지 보장해야 하고, 독립된 지역 또는 국가심의기구로부터 연구 수행에 대한 승인을 받아야 한다고 명시한다. 지침 A.(저자와 기여자의 역할 정의)에서는 교신저자의 의무로 윤리위원회 승인 관련 정보 제공을 명시한다. ICMJE가 준수하는 「헬싱키 선언」은 연구 시작 전 위원회 승인을 받아야 하고(제23조), 선언의 원칙을 위반한 연구는 보고서 출 간이 허용돼서는 안 된다고 규정하므로(제36조), ICMJE도 IRB 승인을 받지 않은 학술 결과의 출 간, 배포, 게재를 허용하지 않는다고 해석할 수 있다. 더불어, 임상시험의 경우 「헬싱키 선언」에 준하여 대상자 모집 전 공적 기관에서 무료로 운영하는 공공 임상시험 레지스트리에 연구 방법과 내용을 등록해야만 논문 게재가 가능하다는 조건을 두고, 초록 말미에 등록번호를 제시하도록 권장한다.
연구 등록 시 대표적으로 활용되는 공공 임상시험 레지스트리는 WHO에서 2007년부터 운영하는 ICTRP(International Clinical Trials Registry Platform)이다. ICTRP는 16개의 국가별 등록 시스템을 구축·운영하고 있으며 미국 NIH의 clinicaltrial.gov와 국내는 질병관리본부의 임상연 구정보서비스(CRIS)가 WHO ICTRP에 포함된다. 이들 데이터베이스는 ‘IRB 심의·승인 정보’를 등록하도록 하여, 연구의 결과 관리 측면에서 IRB 심의를 확인할 수 있는 간접 기제로 활용될 수 있다. 하지만, <Table 2>와 같이 등록 대상이 임상시험에 국한되어 여타 사회과학을 포함한 인간대상연구에 대한 IRB 심의 관리책은 될 수 없다. 또한, IRB 심의 및 승인 정보가 필수가 아닌 곳도 있고 IRB 심의 결과와 정보만 기술하게 한 곳도 있어 연구자가 허위로 IRB 승인 정보를 기재할 가능성을 전면 예방하기에는 충분하지 않을 수 있다.
| 등록 플랫폼 | 대상 | IRB 심의 관련 등록 내용 | 비고 |
|---|---|---|---|
| WHO ICTRP[25] | 전 세계 16개국 국가별 임상시험 등록시스템 통합 운영 | 윤리심의(Ethical Review) 항목에 “심의결과, 승인일, 위원회 정보 및 연락처” 등록 | 필수 아님[26] |
| Clinicaltrial.gov[27,28] | 임상 2상부터 4상 의약품 및 의료기기 임상시험이면서 NIH로부터 연구비 일부 또는 전액 지원 받은 임상시험 | 인간대상심의(Human Subjects Review) 항목에 “승인결과, 승인번호, 위원회 및 기관명, 위원회 연락정보” 등록 | 승인결과(필수), 나머지 정보는 해당 시 필수[29] |
| 질병관리본부 임상연구정보 서비스(CRIS) | 보건복지부 지원 임상연구 만 등록 의무, 그 외 임상 시험은 등록 권고 | 임상연구윤리심의 항목에 “승인상태, 승인번호, 승인일, 승인서 제출(첨부), 위원회 명” 등록 | 승인상태, 번호, 승인서 제출만 필수[26] |
Ⅳ. IRB 심의 미준수 관리 방안 검토
앞서 살펴본 IRB 심의 의무에 대한 국내외 법과 정책적 방안을 바탕으로 우리나라에서 연구자의 IRB 심의 미준수를 예방하고 관리할 수 있는 방안을 검토하고자 한다.
먼저, 연구자에게 직접적 규제나 의무를 부여하기 전에 IRB 심의를 연구자가 자연스럽게 준수하는 환경을 조성하는 예방적 관리 방안이다. 연구자 스스로가 연구 시작 전 기관위원회 심의 의무를 준수하고, 이후 연구의 윤리적 수행을 보장하고 원활히 관리한다는 측면에서 생명윤리법이 지향하는 자율적 규제의 실현에 가장 가까운 관리 방식일 것이다. 이를 위해서는 무엇보다도 연구자, 기관위원회, 기관의 역량 강화가 필수적이다. 연구자는 교육을 통해 IRB 심의를 비롯한 의무에 대해 충분히 숙지하여 이를 준수하도록 해야 하고, 기관위원회는 제출된 연구계획서를 심의하고 조사·감독하여 미준수를 관리할 수 있어야 하며, 기관에서 수행되는 잠재적 심의 대상 연구도 관리 할 수 있어야 한다. 기관위원회가 잠재적 심의 대상 연구를 직접 관리하든, 기관 내 적절한 부서에서 관리하고 기관위원회와 소통하도록 하든, 관리 방식은 기관에서 결정할 수 있겠으나 기관과 기관위원회가 이에 대한 관리의 필요성을 이해하고 준수하도록 하는 것이 필요하다. 또한 기관은 기관위원회 운영을 위한 지원과 기관 내에서 수행되는 연구에 대한 총체적인 책임을 갖도록 해야 한다. 이를 위해서는 미국의 OHRP와 같이 IRB 관리 전담 기구를 통해 IRB의 등록부터 교육 및 질 보 증의 단계를 종합적으로 관리하여, 기관 내 각 주 체별 역량이 강화될 수 있도록 IRB를 지원하고 관리하는 방안이 필요하다.
우선, 법률의 적용 범위와 대상을 명확하게 할 필요가 있다. 현재 생명윤리법은 기관위원회 설치 및 등록 의무를 심의 대상에 속하는 ‘연구를 수행하려는 자가 속한 기관’으로 규정하고, 그 기관도 병원, 대학 및 연구기관 등으로 매우 포괄적이다. 반면, 설치 예외 규정 없이 위탁 또는 협약으로 할 수 있는 기준만 제시하고 있다. 따라서 해당 연구를 수행하려는 연구자가 1명이라도 있다면, 원칙적으로 IRB를 설치 및 등록해야 하고 미준수 시 과태료 대상이 된다. 그러나 과연 1명의 연구자를 위한 연구를 위해 기관장이 기관위원회 운영을 협약 또는 설치해야 한다고 강제하는 것이 적절한지에 대해서는 재고가 필요하다. 오히려 이런 경우에는 설치 예외 대상을 규정하고, 대안으로 선택할 수 있는 공용 IRB 등을 지원하는 방안이 필요할 것이다. 또한 IRB의 적절한 운영을 위한 독립성과 전문성을 위한 요건으로 적절한 운영의 규모를 제시하고 그 운영의 요건을 만족하지 않을 것이라고 예상되는 기관에게 협약 기준을 적용하는 것이 바람직할 것이다. 따라서 IRB 설치 대상과 예외에 대한 명확한 기준, 그리고 운영이 적절하지 않은 경우에 대한 협약 기준으로 구분되어 제시되어야 할 것이다. 이러한 설치 대상에 대한 명확한 근거가 마련되면, 이를 바탕으로 IRB 등록 단계에서 IRB 관리 기관은 등록 신청 기관의 인프라와 연구 규모 등을 고려하여 해당 기관이 직접 기관위원회를 운영할 수 있는 역량이 있는지, 위탁이나 협약에 따른 운영이 더 적절할지에 대한 컨설팅을 제공할 수 있을 것이다. 기관에 맞는 적절한 컨설팅을 제공하기 위해 전문기관의 역량 확보와 컨설팅의 표준화 등도 필요할 것이며, IRB가 설치되지 않은 기관의 연구자에게 공용 IRB 심의 요청 근거를 마련하는 등 공용 IRB의 운영 확대와 활성화를 통한 지원 정책도 고려되어야 할 것이다.
또한 미국과 같이 등록 의무 및 준수 의무의 대상을 기관으로 하고, IRB 심의를 포함한 법규 준수 보장에 대한 준수 서약을 제출하도록 하여 기관의 책임을 강조하여 관리하는 방안도 고려될 수 있다. 생명윤리법은 기관위원회의 구성과 운영을 위해 기관장에게 행정적·재정적 지원을 하도록 하고 있으나, 구체적인 지원의 범위나 기준 또는 책임을 표준화하기는 매우 어렵기 때문에 적절한 관리나 지원을 보장하기도 쉽지 않다. 그러나 운영지원에 실질적인 기여를 하는 요소이므로 적절한 관리는 매우 중요하다. 따라서 미국과 같이 기관에 심의 준수와 IRB 운영에 대한 법적 구속력과 책임을 부여하는 서약과 같이, 기관의 책무를 강조하는 제도적 장치를 통하여 기관 내 연구나 IRB 운영에 대하여 적극적인 개입을 독려하는 방안도 고려해 볼 수 있다.
다음은 교육의 일원화가 필요하다. 교육의 실효 성에 대한 논란도 있으나 그렇기에 교육 단계에서의 체계화된 기획과 교육의 시행 및 평가 등이 중요하다. 연구자는 교육을 통해 연구 수행 전 IRB 심의에 대한 의무와 책임을 알고 이를 실천하도록 지원받아야 한다. 임상시험 연구자는 1995년 부터 시행된 「임상시험관리기준」과 공공 임상시험 레지스트리 등록 의무를 통해 IRB 심의 의무에 많이 노출되어 있던 반면, 인문사회 및 사회과학 연구자는 IRB에 대한 이해와 경험이 상대적으로 부족하다. 따라서 이들 연구자의 연구 수행 범위를 고려하는 동시에 연구의 윤리적 수행에 대한 역할과 의무를 배양할 수 있는 심층적 교육 모듈이 개발 및 제공되어야 한다. IRB 관리를 전담하는 기구는 기관위원회 등록 시 기관이 제출한 기본 자 료와 기관위원회 운영 실태 등에 대한 조사(법 제13조 및 시행규칙 제12조), 그리고 기관위원회 평가·인증 등을 통해 기관위원회 운영 현황과 해당 기관위원회의 역할 범주 및 연구대상을 고려한 현실적인 교육을 제공해야 한다. 교육 방식 또한 일방적인 주입식 교육보다는, 해당 기관에 실질적으로 필요한 역량을 지원할 수 있는 방식으로 제공되어야 한다. 마지막으로 기관위원회 운영의 질을 보증하는 단계에서는 관리 효과가 나타날 수 있어야 한다. 기관 내 연구 부처와 협업하여 연구자의 연구 실적을 파악하고 IRB 심의가 준수되었는지 관리하는 체계가 지원되고 점검되어야 한다. 또한, IRB에는 미준수와 관련한 기관위원회의 조사·감독 과정, 절차, 조치를 관리기구에 보고하도록 하고, 해당 기구는 이에 대한 교육 및 컨설팅을 제공하여 적절한 시정 조치를 할 수 있도록 지원해야 한다. 이처럼 기관위원회와 IRB 관리기구가 조사·감독 과정에서 함께 협력한다면, 조사· 감독 미준수에 따른 후속 조치를 집행하고 관리하는 등 기관에서 실제 기능하고 위상을 갖추게 될 것이다.
IRB에 대한 이러한 종합적이고 통합적인 관리는 궁극적으로 기관에게는 IRB 운영을 위한 지원과 연구의 윤리적 수행을 보장하는 능력을 배양하도록 하고, 연구자에게는 연구와 관련한 의무와 책임을 인지하고 준수하도록 하며, IRB에게는 기관 내 생명·안전·윤리 관리 조직이라는 위상을 부여하고 연구 행위 전반을 관리·감독하도록 할 것이다. 결과적으로 IRB 심의 미준수와 같은 위반 행위를 스스로 예방할 수 있는 역량을 갖추도록 한다. 다만, 기관위원회 전담 관리 기구의 설치와 운영은 별도의 기구를 설치할지, 기관위원회 관리 담당 기관 중 현재의 기능을 확대하여 운영할지에 대한 고려가 필요하며 등록·교육·질 관리를 위한 전문 인력과 자원의 확보가 절대적으로 요구된다. 따라서 기관위원회를 관리하는 보건복지부와 관련 부처 등의 정부가 기관위원회의 역량 강화와 통합적, 체계적, 효율적 관리에 얼마나 확고한 의지가 있는지에 따라 고려가 가능할 것이다.
연구를 지원·관리하는 기관과 학술지를 관리하는 학술기관은 기관에서 정한 기준과 원칙에 따라 연구의 윤리적 수행 여부를 확인하며, 이때 IRB 심의 여부를 표기하거나 승인번호를 제출하는 방식을 주로 사용한다. 한국연구재단도 IRB 심의를 의무로 정하고 있으나, 해당 연구가 심의가 필요한지에 대한 객관적 판단에 대한 책임 없이, 연구비 신청 단계에서는 신청서에 연구자가 IRB 심의를 받았다는 체크를 확인하고, 연구 종료 후 결과 보고 시에는 심의 대상인 경우 IRB 심의 결과서를 첨부하도록 하고 있다[30]. 하지만 심의대상이나 연구자가 임의로 심의대상이 아니라고 판단했다면 이를 확인하여 제재할 방법은 여전히 없으므로 심의 미준수 관리가 적절하게 되고 있다고 보기에는 한계가 있어 보인다.
미국 NIH의 경우 연구지원을 신청하는 단계에서 심사 승인서를 요구하거나 IRB 질 보증의 근거를 제출하도록 요구하고 충족 여부를 검토하여 지원 여부를 결정한다. 다만, 연구자에 대한 사전 심의 승인서 제출을 강제할 경우에는 실제 제출할 수 있는 가능성이 매우 낮다. 따라서 이 경우는 승 인서 대신 심사 신청서로 대체하거나, 심의 확인을 조건으로 연구를 수행하도록 강제하는 방법도 고려될 수 있겠다.
일반적으로 새로운 과학기술 발전과 생명과 학·의학 등 인간을 대상으로 한 연구에서 발생 가능한 윤리적 문제를 다루는 생명윤리8)와 연구 과정에서의 결과 활용, 부정행위 및 연구진실성에 대한 연구윤리는 따로 관리된다. 국내의 경우도 생명윤리는 생명윤리법에 근거하여 기관생명 윤리위원회에서 관리하고, 연구윤리는 학술진흥 법 내지는 국가연구개발사업의 관리 등에 관한 규정에 따라 연구진실성조사위원회에서 관리하고 있다[32]. 하지만 연구대상자의 권리와 안전보호는 생명윤리와 연구윤리의 공통 영역이라는 점을 감안할 때[33]. 영국 COPE와 같이 연구부정행위에 IRB 미심의와 승인 허위기재를 포함하여 관리하는 방안도 참고할 수 있을 것이다. 또한, NIH 처럼 연구 중간점검 과정에서 연구의 윤리적 수행과 안전성을 확인하거나, 「정책연구 용역과제 관리지침」 제20조에 따라 연구비 지급 보류 또는 중단이 가능한 조건인 ‘연구 진행 상황보고를 허위로 한 경우’ 및 ‘연구 부정행위’와 같은 범위에, 이미 제출된 신청서상 IRB 심의와 승인 여부를 허위 기재한 경우를 포함하여 관리하는 방안도 고려될 수 있다. 마지막으로, 학술기관은 ICMJE와 같이 인간대상연구를 수행한 교신저자로부터 기관위원회 승인 정보를 제공하도록 하고 직접 해당 사실을 확인한 후에 게재를 허용하는 방안도 검토할 수 있다. 이러한 정책은 재단으로부터 연구비를 지원 받는 연구와 학술기관에 논문을 투고한 경우에만 적용되어 관리 대상이 제한적일 수는 있으나, 이를 계기로 IRB 심의 의무가 준수되는 연구 환경과 기반이 마련되고 그 범위가 점차 확대될 수 있을 것이다.
세계의사회의 「헬싱키 선언」과 ICMJE의 「의학 학술지 학술 업적의 수행, 보고, 편집, 발간에 관한 권고」 지침은 임상시험의 경우 대상자 모집 전 IRB 승인 정보 및 연구 실적을 공공 시스템에 등록하도록 권고하고 있다. 이러한 등록 시스템은 근거중심의학 개념의 확산에 따라 임상시험의 객관성 확보와 임상결과의 선택적 보고 또는 출판 비틀림(bias)으로부터 연구대상자를 보호하기 위한 목적으로 마련되었으나, 연구의 투명성 확보와 윤리적 수행을 가능하게 하는 기제로 두루 활용되고 있다. 따라서 IRB 심의 미준수에 대한 객관성 보장과 투명성 확보에 대한 요구가 있을 때는 연구 실적과 IRB 승인 정보를 제공하는 시스템을 고려해볼 수 있다. 해당 시스템은 생명윤리법에 적용되는 모든 연구 실적의 파악과 이들 연구가 법에 따라 IRB 심의를 받았는지를 확인할 수 있어야 한다. 따라서 기존 시스템과 같이 연구자가 모든 정보를 입력하도록 하기보다는, 연구자와 기관위원회가 각자 해당 정보를 등록하는 방안이 고려 될 수 있다. 예컨대, 연구자에게는 계획 중인 연구 정보를 등록하도록 하고, 다음으로는 기관위원회에 IRB 승인 정보를 입력하도록 한 후, 시스템 관리자가, 연구자가 등록한 연구정보와 기관위원회가 등록한 IRB 승인정보를 연계하는 방안이 있을 것이다. 계획 및 수행 중인 연구자의 모든 연구 실적을 파악할 수 있고 IRB에서 공식적인 승인 정보를 제공하므로, IRB 승인의 투명성이 보장되며 허위 기재를 예방할 수 있다.
연구에 대한 등록과 정보 공유는 연구자나 기관위원회의 운영에 불필요한 부담이 발생하지 않는 적정 수준에서 목적에 부합하게 고려되어야 한다. 연구의 IRB 심의와 승인 정보 확인이 목적이므로 모두에게 최소한의 필수 정보만 선별하여 등록하도록 할 필요가 있다. 현재 대부분의 기관위원회가 IRB 심의 및 승인 정보를 엑셀 또는 e-IRB를 통해 관리하고 있으므로, e-IRB 시스템에서 등록에 필요한 정보를 추출할 수 있도록 하거나 표 준화된 엑셀 파일 업로드만으로 등록이 가능하도록 하는 등의 편리성이 고려되어야 한다. 또한, 시스템 관리자가 연구자 및 기관위원회 정보를 연계할 때 일정 번호를 자동 생성하도록 하여 연구비 지원이나 연구결과 투고 시 통일된 형식의 번호를 기재하도록 하고 이를 관련자가 적절히 확인하는 방식도 고려될 수 있다.
국내 연구 등록을 위해 사용되는 시스템은 한국 연구재단의 KRI(한국연구자정보시스템)와 질병 관리본부의 CRIS(임상연구정보서비스)가 대표적이며, 두 시스템 모두 연구자만 등록할 수 있다. KRI는 학술지에 게재된 연구 업적을 등록하도록 하여 아직 시작하지 않거나 계획 중인 연구는 파악하기 어렵고, CRIS는 정부 지원을 받은 임상연 구에 한정되며 연구 유형과 관계없이 세부 정보까지 등록하도록 요구하여 두 시스템 모두 그대로 이용하기에는 제약이 있다. 따라서 기존 시스템을 보강하거나, IRB 관리 기구 또는 생명윤리법을 관할하는 보건복지부와 같은 정부기관이 목적에 맞는 새로운 시스템을 운영하는 방안이 고려되어야 할 것으로 보인다. 다만, 시스템을 통한 IRB 승인과 허위기재 관리는 연구 커뮤니티로부터 수용 이 매우 중요한 사항이 될 것이고, 예산과 운영 방법에 대한 면밀한 검토가 필요하므로 IRB 심의를 준수하지 않은 비윤리적 연구에 대한 정보 공유와 객관성·투명성 확보에 대해 사회적 합의가 이루어진 경우에 재고가 가능할 것이다.
V. 마치며
본 글은 책임 있는 연구의 시작점인 IRB 심의를 중점으로 연구 수행과 관리에 대한 생명윤리법의 취지와 그에 따른 한계를 살펴보고, 미국을 중심으로 국내외 IRB 심의에 대한 법률과 정책을 검토하여 심의 관련 미준수를 관리할 수 있는 방안을 모색했다. 특히, <Figure 1>과 같이 국내에 적용 가능한 IRB 심의 관리 방안으로 IRB 관리기구를 통한 교육·역량 강화와 같은 ‘예방적 관리’, 연구 및 학술 관리 기구를 통한 ‘정책적 관리’, 그리고 연구 및 승인 정보의 등록과 공유를 통한 ‘시스템적 관리’를 살펴보았다.
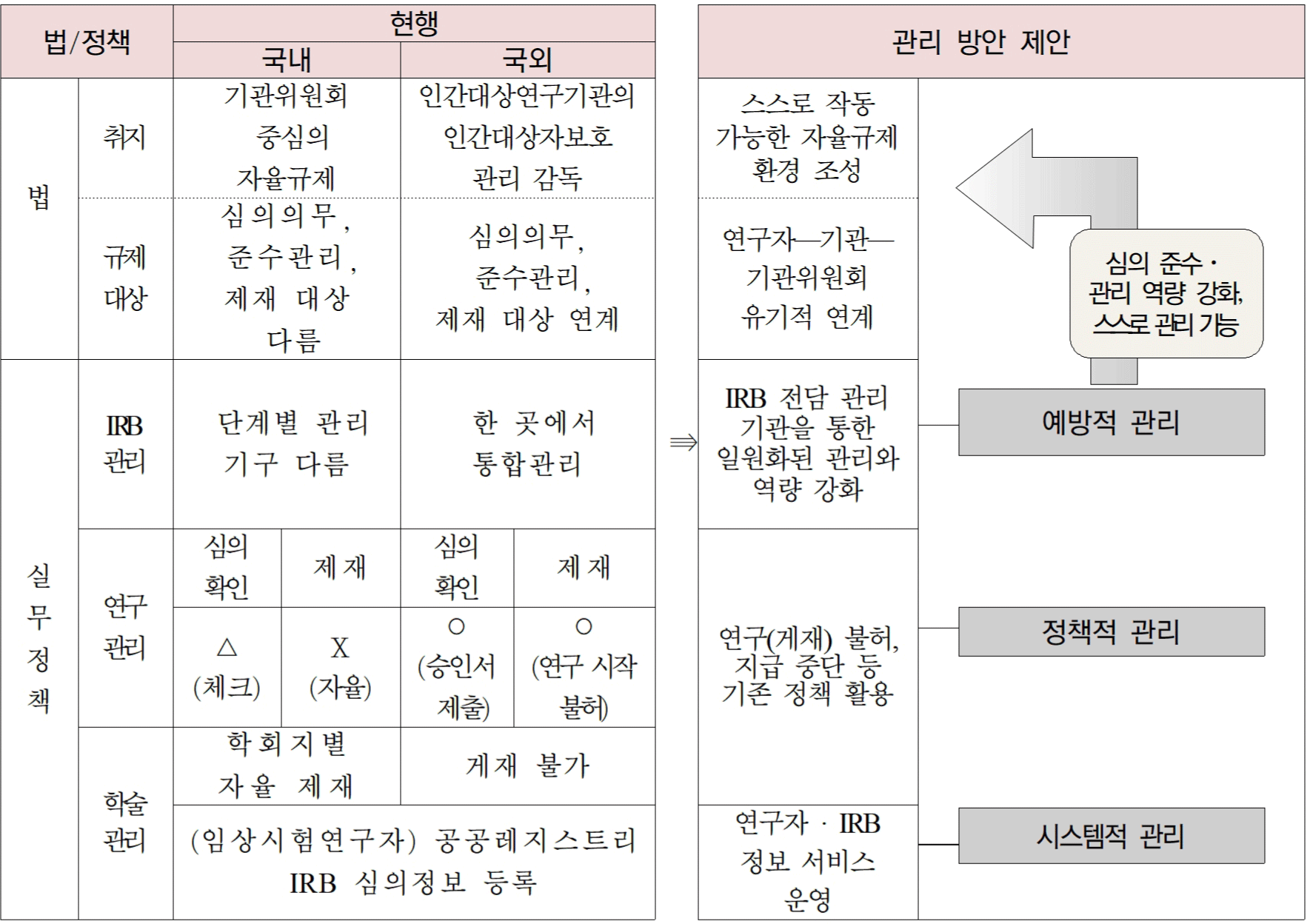
IRB 심의 준수는 연구자가 연구를 수행하면서 지켜야할 기본 행동 양식이며 윤리성에 기반한다. 따라서 제재를 동반한 정책적 관리나 사실 확인을 위한 시스템적 관리보다는, IRB 관리 기구를 주축으로 기관·기관위원회·연구자 모두의 역량을 강화하는 교육과 제도의 보완을 통한 예방적 관리가 무엇보다 중요하겠다.
국내에서 연구에서의 생명윤리와 안전은 생명 윤리법을 통해 확보된다. 2012년 전부 개정된 생명윤리법은 인간과 인체유래물을 연구하거나 배아나 유전자 등을 취급할 때 인간의 존엄과 가치를 침해하거나 인체에 위해를 끼치는 것을 방지하는 시스템을 충실히 갖추었다. 예방적 차원의 법적 체계는 건실한 데 반해, 미준수에 대한 관리 시스템은 상대적으로 부족하다. 특히 대부분이 IRB 심의를 받고 수행 중인 연구 관리에 초점을 두어, IRB 심의 미준수와 같이 연구 출발부터 잘못된 사례에 대한 관리는 더욱 부족하다. 또한 연구자의 IRB 심의 준수 여부를 기관위원회 평가·인증을 통해 관리하고, 인증결과에 따라 제재를 부여할 수 있도록 정하고 있어, 향후 기관위원회 인증 제도의 체계 구축과 관리 방안이 무엇보다도 중요 할 것이다. IRB 심의는 의례적인 절차가 아닌 연구의 윤리적 시작과 진행 및 종료를 담보할 수 있으므로 미준수에 대한 체계적인 점검과 관리 방안의 마련은 중요하다. 본 글에서 검토한 방안이 향후 IRB 심의 준수 관리뿐 아니라 기관위원회 인증 제도와 생명윤리 및 안전 관리 체계를 적절한 방향으로 확립하는 데 도움이 되기를 바란다.